hBayesDM (hierarchical Bayesian modeling of Decision-Making tasks) is a user-friendly R package that offers hierarchical Bayesian analysis of various computational models on an array of decision-making tasks. Click here to download its help file (reference manual). Click here to read our paper published in Computational Psychiatry. Click here to download a poster we presented at several conferences/meetings. You can find hBayesDM on CRAN and GitHub.
Motivation
Computational modeling provides a quantitative framework for investigating latent neurocognitive processes (e.g., learning rate, reward sensitivity) and interactions among multiple decision-making systems. Parameters of a computational model reflect psychologically meaningful individual differences: thus, getting accurate parameter estimates of a computational model is critical to improving the interpretation of its findings. Hierarchical Bayesian analysis (HBA) is regarded as the gold standard for parameter estimation, especially when the amount of information from each participant is small (see below “Why hierarchical Bayesian analysis?”). However, many researchers interested in HBA often find the approach too technical and challenging to be implemented.
We introduce a free R package hBayesDM, which offers HBA of various computational models on an array of decision-making tasks (see below for a list of tasks and models currently available). Users can perform HBA of various computational models with a single line of coding. Example datasets are also available. With hBayesDM, we hope anyone with minimal knowledge of programming can take advantage of advanced computational modeling and HBA. It is our expectation that hBayesDM will contribute to the dissemination of these computational tools and enable researchers in related fields to easily characterize latent neurocognitive processes within their study populations.
Why hierarchical Bayesian analysis (HBA)?
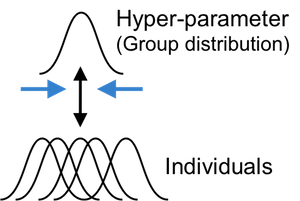
Most computational models do not have closed form solutions and we need to estimate parameter values. Traditionally parameters are estimated at the individual level with maximum likelihood estimation (MLE): getting point estimates for each individual separately. However, individual MLE estimates are often noisy especially when there is insufficient amount of data. A group-level analysis (e.g., group-level MLE), which estimate a single set of parameters for the whole group of individuals, may generate more reliable estimates but inevitably ignores individual differences.
HBA and other hierarchical approaches (e.g., Huys et al., 2011) allow for individual differences while pooling information across individuals. Both individual and group parameter estimates (i.e., posterior distributions) are estimated simultaneously in a mutually constraining fashion. Consequently, individual parameter estimates tend to be more stable and reliable because commonalities among individuals are captured and informed by the group tendencies (e.g., Ahn et al., 2011). HBA also finds full posterior distributions instead of point estimates (thus providing rich information about the parameters). HBA also makes it easy to do group comparisons in a Bayesian fashion (e.g., comparing clinical and non-clinical groups, see an example below).
HBA is a branch of Bayesian statistics and the conceptual framework of Bayesian data analysis is clearly written in Chapter 2 of John Kruschke’s book (Kruschke, 2014). In Bayesian statistics, we assume prior beliefs (i.e., prior distributions) for model parameters and update the priors into posterior distributions given the data (e.g., trial-by-trial choices and outcomes) using Bayes’ rule. Note that the prior distributions we use for model parameters are vague (e.g., flat) or weakly informative priors, so they play a minimal role in the posterior distribution.
For Bayesian updating, we use the Stan software package (https://mc-stan.org/), which implements a very efficient Markov Chain Monte Carlo (MCMC) algorithm called Hamiltonian Monte Carlo (HMC). HMC is known to be effective and works well even for large complex models. See Stan reference manual (https://mc-stan.org/documentation/) and Chapter 14 of Kruschke (2014) for a comprehensive description of HMC and Stan. What is MCMC and why should we use it? Remember, we need to update our priors into posterior distributions in order to make inference about model parameters. Simply put, MCMC is a way of approximating a posterior distribution by drawing a large number of samples from it. MCMC algorithms are used when posterior distributions cannot be analytically achieved or using MCMC is more efficient than searching for the whole grid of parameter space (i.e., grid search). To learn more about the basic foundations of MCMC, we recommend Chapter 7 of Kruschke (2014).
Users can read our Stan source files to see how each model is
implemented (e.g.,
pathTo_gng_m1 = system.file("stan_files/gng_m1.stan", package="hBayesDM")).
We made strong efforts to optimize Stan codes through reparameterization
(e.g., Matt trick) and vectorization.
Prerequisites
- R ≥ 4.4 (cran.r-project.org).
-
CmdStan, installed via cmdstanr.
hBayesDM uses cmdstanr (not rstan) to compile and run Stan models.
CmdStan ships as a system dependency rather than a CRAN package —
install it once with
cmdstanr::install_cmdstan(). - RStudio is optional but recommended.
Additional CRAN packages (ggplot2, loo,
bayesplot, posterior, …) install automatically
with hBayesDM.
Tasks & models implemented in hBayesDM
See here for the list of tasks and models implemented in hBayesDM.
How to install hBayesDM
1. Install cmdstanr and CmdStan
cmdstanr is hosted on the Stan r-universe rather than CRAN:
install.packages(
"cmdstanr",
repos = c("https://stan-dev.r-universe.dev", getOption("repos"))
)
# One-time CmdStan toolchain install (~5 min)
cmdstanr::install_cmdstan()Verify the toolchain works by fitting any Stan model — for example,
cmdstanr::cmdstanr_example("schools") runs a small “Eight
Schools” fit without hBayesDM.
2. Install hBayesDM
From CRAN:
install.packages("hBayesDM")From GitHub:
if (!require(remotes)) install.packages("remotes")
remotes::install_github("CCS-Lab/hBayesDM", subdir = "R")How to use hBayesDM
First, open RStudio (or just R) and load the package:
Four steps of doing HBA with hBayesDM are illustrated below. As an example, four models of the orthogonalized Go/Nogo task (Guitart-Masip et al., 2012; Cavanagh et al., 2013) are fit and compared with the hBayesDM package.
1) Prepare the data
- For fitting a model with hBayesDM, all subjects’ data should be
combined into a single text file (*.txt). Look at the sample dataset and
a help file (e.g.,
?gng_m1) for each task and carefully follow the instructions. - Subjects’ data must contain variables that are consistent with the column names specified in the help file, though extra variables are in practice allowed.
- It is okay if the number of trials is different across subjects. But
there should exist no N/A data. If some trials contain N/A data (e.g.,
choice=NAin trial#10), remove the trials first. - Sample data are available here,
although users can fit a model with sample data without separately
downloading them with one of the function arguments. Once the hBayesDM
package is installed, sample data can be also retrieved from the package
folder. Note that the file name of sample (example) data for a given
task is taskName_exampleData.txt (e.g.,
dd_exampleData.txt, igt_exampleData.txt, gng_exampleData.txt, etc.). See
each model’s help file (e.g.,
?gng_m1) to check required data columns and their labels.
dataPath = system.file("extdata/gng_exampleData.txt", package="hBayesDM")If you download the sample data to “~/Downloads”, you may specify the path to the data file like this:
dataPath = "~/Downloads/gng_exampleData.txt"2) Fit candidate models
Below the gng_m1 model is fit on its example data. The
command runs four MCMC chains across four cores. If you pass
"example" as data, hBayesDM loads the bundled
example file for that task. You can also assign your own initial values
via the inits argument (e.g.,
inits = c(0.1, 0.2, 1.0)).
output1 = gng_m1(data="example", niter=2000, nwarmup=1000, nchain=4, ncore=4)Equivalent shorter form (the defaults are 2000 / 1000 / 4 chains for
gng models):
output1 = gng_m1("example", ncore=4)## Warning in gng_m1(data = "example", niter = 2000, nwarmup = 1000, nchain = 4, : Number of cores specified for parallel computing greater than number of locally available cores. Using all locally available cores.##
## Model name = gng_m1
## Data file = example
##
## Details:
## # of chains = 4
## # of cores used = 3
## # of MCMC samples (per chain) = 2000
## # of burn-in samples = 1000
## # of subjects = 10
## # of (max) trials per subject = 240
##
##
## ****************************************
## ** Use VB estimates as initial values **
## ****************************************
## ------------------------------------------------------------
## EXPERIMENTAL ALGORITHM:
## This procedure has not been thoroughly tested and may be unstable
## or buggy. The interface is subject to change.
## ------------------------------------------------------------
## Gradient evaluation took 0.000601 seconds
## 1000 transitions using 10 leapfrog steps per transition would take 6.01 seconds.
## Adjust your expectations accordingly!
## Begin eta adaptation.
## Iteration: 1 / 250 [ 0%] (Adaptation)
## Iteration: 50 / 250 [ 20%] (Adaptation)
## Iteration: 100 / 250 [ 40%] (Adaptation)
## Iteration: 150 / 250 [ 60%] (Adaptation)
## Iteration: 200 / 250 [ 80%] (Adaptation)
## Success! Found best value [eta = 1] earlier than expected.
## Begin stochastic gradient ascent.
## iter ELBO delta_ELBO_mean delta_ELBO_med notes
## 100 -846.796 1.000 1.000
## 200 -815.045 0.519 1.000
## 300 -809.962 0.348 0.039
## 400 -809.490 0.261 0.039
## 500 -810.612 0.209 0.006 MEDIAN ELBO CONVERGED
## Drawing a sample of size 1000 from the approximate posterior...
## COMPLETED.
## Finished in 3.4 seconds.
##
## ************************************
## **** Model fitting is complete! ****
## ************************************On first use cmdstanr compiles the Stan model (~30 s); subsequent
fits of the same model reuse the cached binary. Sampling for
gng_m1 on the example data then takes 2–3 minutes.
Occasional “numerical problems” warnings during warmup or a small number
of “divergent transitions after warmup” can usually be ignored — see
Stan’s Runtime warnings
and convergence problems for guidance on when to act on them.
The hBayesDM console output looks like:
Model = gng_m1
Data = example
Details:
# of chains = 4
# of cores used = 4
# of MCMC samples (per chain) = 2000
# of burn-in samples = 1000
# of subjects = 10
# of (max) trials per subject = 240
Running MCMC with 4 parallel chains...
Chain 1 Iteration: 1 / 2000 [ 0%] (Warmup)
Chain 2 Iteration: 1 / 2000 [ 0%] (Warmup)
...When fitting is complete:
************************************
**** Model fitting is complete! ****
************************************output1 is an object of class hBayesDM with
these fields:
-
model: Name of the fitted model (output1$modelis"gng_m1"). -
all_ind_pars: Per-subject parameter summaries (default: mean). Useind_pars = "median"orind_pars = "mode"to change the summary statistic. -
par_vals: Posterior draws as a named list, extracted viaposterior::draws_of(). Hyper-parameters are prefixedmu_(e.g.,mu_xi,mu_ep,mu_rho). -
fit: ACmdStanMCMCobject (orCmdStanVBwhenvb = TRUE) from cmdstanr. Useoutput1$fit$summary(),output1$fit$draws(),output1$fit$diagnostic_summary(), etc. — see?cmdstanr::CmdStanMCMC. -
raw_data: Raw trial-by-trial data used for modeling. Provided so users can easily compare trial-by-trial model-based regressors (e.g., prediction errors) against the choice data. -
model_regressor(optional): Trial-by-trial model-based regressors such as prediction errors and chosen-option values. Each model ships with a pre-selected set.
> output1$all_ind_pars
xi ep rho subjID
1 0.03688558 0.1397615 5.902901 1
2 0.02934812 0.1653435 6.066120 2
3 0.04467025 0.1268796 5.898099 3
4 0.02103926 0.1499842 6.185020 4
5 0.02620808 0.1498962 6.081908 5
...Use output1$fit$summary() to get a posterior summary
table:
> output1$fit$summary(c("mu_xi", "mu_ep", "mu_rho"))
# A tibble: 3 × 10
variable mean median sd mad q5 q95 rhat ess_bulk ess_tail
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 mu_xi 0.03 0.03 0.02 0.02 0.00 0.07 1.00 2316 2780
2 mu_ep 0.15 0.15 0.02 0.02 0.11 0.19 1.00 4402 3120
3 mu_rho 5.97 5.89 0.72 0.71 4.76 7.61 1.00 3821 3055Or call rhat(output1) for an hBayesDM-friendly Rhat
data.frame (see Section 3 below).
\(\hat{R}\) (Rhat) is
an index of MCMC convergence. Values close to 1.00 indicate that the
chains have converged to a stationary target distribution. On hBayesDM
example data, most parameters reach \(\hat{R}
\le 1.01\).
3) Plot model parameters
Always visually diagnose MCMC performance (well-mixed, stationary
chains) before interpreting parameter estimates. For hyper-parameters,
use plot.hBayesDM (or just plot — S3 dispatch
picks it up since the result inherits from hBayesDM).
Plotting is backed by bayesplot.
Trace plots of the hyper-parameters:
plot(output1, type="trace") # traceplot via bayesplot::mcmc_trace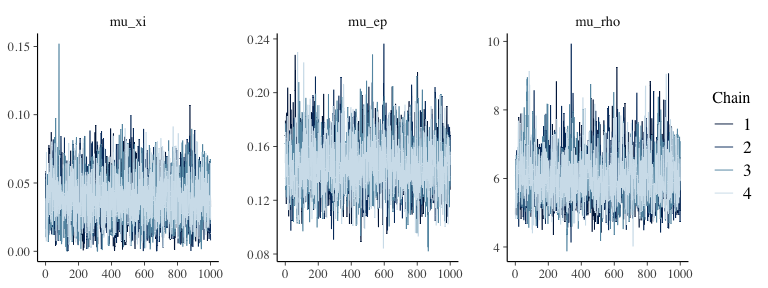
Well-mixed traces look like “fuzzy caterpillars” and confirm what the \(\hat{R}\) values suggest (see this StackExchange thread for why mixing matters). Trace plots show post-warmup draws only.
For posterior interval / density plots of the hyper-parameters:
plot(output1, type="simple") # bayesplot::mcmc_intervals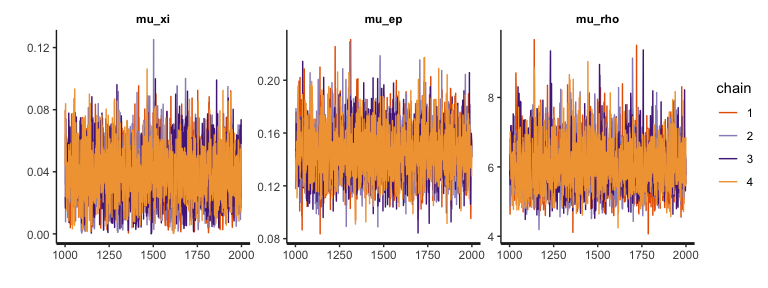
plot(output1) # type="dist" — per-model density plots## Warning: The dot-dot notation (`..density..`) was deprecated in ggplot2 3.4.0.
## ℹ Please use `after_stat(density)` instead.
## ℹ The deprecated feature was likely used in the hBayesDM package.
## Please report the issue at <https://github.com/CCS-Lab/hBayesDM/issues>.
## This warning is displayed once per session.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.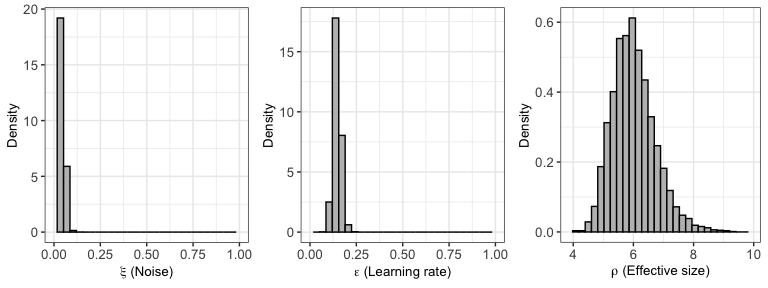
To inspect individual subjects, use plot_ind() (built on
bayesplot::mcmc_areas / mcmc_intervals). For
example, each subject’s \(\epsilon\)
(learning rate) posterior:
plot_ind(output1, "ep")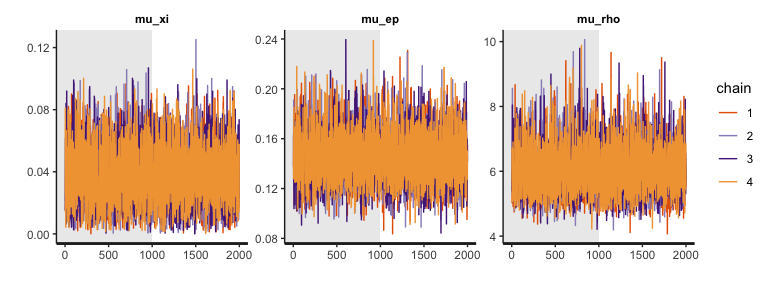
You can also call rhat(output1) to get a data.frame of
Rhat values, or rhat(output1, less = 1.05) to get a
TRUE/FALSE summary check.
4) Compare models (and groups)
To compare models, you first fit all models in the same manner as the
example above (e.g.,
output4 = gng_m4("example", niter=2000, nwarmup=1000, nchain=4, ncore=4)
). Next, we use the command print_fit, which is a
convenient way to summarize Leave-One-Out Information Criterion (LOOIC)
or Widely Applicable Information Criterion (WAIC) of all models we
consider (see Vehtari et al. (2015) for
the details of LOOIC and WAIC). By default, print_fit
function uses the LOOIC which is preferable to the WAIC when there are
influential observations (Vehtari et al.,
2015).
Assuming four models’ outputs are output1 (gng_m1),
output2 (gng_m2), output3 (gng_m3), and
output4 (gng_m4), their model fits can be simultaneously
summarized by:
> print_fit(output1, output2, output3, output4)
Model LOOIC
1 gng_m1 1588.843
2 gng_m2 1571.129
3 gng_m3 1573.872
4 gng_m4 1543.335Note that the lower LOOIC is, the better its model-fit is. Thus,
model#4 has the best LOOIC compared to other models. Users can print
WAIC or both by calling
print_fit(output1, output2, output3, output4, ic="waic") or
print_fit(output1, output2, output3, output4, ic="both").
Use the extract_ic function (e.g.,
extract_ic(output3) ) if you want more detailed information
including standard errors and expected log pointwise predictive density
(elpd). Note that the extract_ic function can be used only
for a single model output.
We also want to remind you that there are multiple ways to compare computational models (e.g., simulation method (absolute model performance), parameter recovery, generalization criterion) and the goodness of fit (e.g., LOOIC or WAIC) is just one of them. Check if predictions from your model (e.g., “posterior predictive check”) can mimic the data (same data or new data) with reasonable accuracy. See Kruschke (2014) (for posterior predictive check), Guitart-Masip et al. (2012) (for goodness of fit and simulation performance on the orthogonalized Go/Nogo task), and Busemeyer & Wang (2000) (for generalization criterion) as well as Ahn et al. (2008; 2014) and Steingroever et al. (2014) (for the combination of multiple model comparison methods).
To compare two groups in a Bayesian fashion (e.g., Ahn et al., 2014), first you need to fit each group with the same model and ideally the same number of MCMC samples. For example,
data_group1 = "~/Project_folder/gng_data_group1.txt" # data file for group1
data_group2 = "~/Project_folder/gng_data_group2.txt" # data file for group2
output_group1 = gng_m4(data_group1) # fit group1 data with the gng_m4 model
output_group2 = gng_m4(data_group2) # fit group2 data with the gng_m4 model
## After model fitting is complete for both groups,
## evaluate the group difference (e.g., on the 'pi' parameter) by examining the posterior distribution of group mean differences.
diffDist = output_group1$par_vals$mu_pi - output_group2$par_vals$mu_pi # group1 - group2
hdi( diffDist ) # Compute the 95% Highest Density Interval (HDI).
plot_hdi( diffDist ) # plot the group mean differences5) Extracting trial-by-trial regressors for model-based fMRI/EEG analysis
In model-based neuroimaging (e.g., O’Doherty et al., 2007), model-based time series of a latent cognitive process are generated by computational models, and then time series data are convolved with a hemodynamic response function and regressed again fMRI or EEG data. This model-based neuroimaging approach has been particularly popular in cognitive neuroscience.
The biggest challenge for performing model-based fMRI/EEG is to learn how to extract trial-by-trial model-based regressors. The hBayesDM package allows users to easily extract model-based regressors that can be used for model-based fMRI or EEG analysis. The hBayesDM package currently provides the following model-based regressors. With the trial-by-trial regressors, users can easily use their favorite neuroimaging package (e.g., in Statistical Parametric Mapping (SPM; http://www.fil.ion.ucl.ac.uk/spm/) to perform model-based fMRI analysis. See our paper (Extracting Trial-by-Trial Regressors for Model-Based fMRI/EEG Analysis) for more details.
As an example, if you would like to extract trial-by-trial stimulus
values (i.e., expected value of stimulus on each trial), first fit a
model like the following (set the model_regressor input
variable to TRUE. Its default value is
FALSE):
## fit example data with the gng_m3 model
output3 = gng_m3(data="example", niter=2000, nwarmup=1000, model_regressor=TRUE)## Warning in gng_m3(data = "example", niter = 2000, nwarmup = 1000, nchain = 4, : Number of cores specified for parallel computing greater than number of locally available cores. Using all locally available cores.##
## Model name = gng_m3
## Data file = example
##
## Details:
## # of chains = 4
## # of cores used = 3
## # of MCMC samples (per chain) = 2000
## # of burn-in samples = 1000
## # of subjects = 10
## # of (max) trials per subject = 240
##
## **************************************
## ** Extract model-based regressors **
## **************************************
##
##
## ****************************************
## ** Use VB estimates as initial values **
## ****************************************
## ------------------------------------------------------------
## EXPERIMENTAL ALGORITHM:
## This procedure has not been thoroughly tested and may be unstable
## or buggy. The interface is subject to change.
## ------------------------------------------------------------
## Gradient evaluation took 0.000695 seconds
## 1000 transitions using 10 leapfrog steps per transition would take 6.95 seconds.
## Adjust your expectations accordingly!
## Begin eta adaptation.
## Iteration: 1 / 250 [ 0%] (Adaptation)
## Iteration: 50 / 250 [ 20%] (Adaptation)
## Iteration: 100 / 250 [ 40%] (Adaptation)
## Iteration: 150 / 250 [ 60%] (Adaptation)
## Iteration: 200 / 250 [ 80%] (Adaptation)
## Success! Found best value [eta = 1] earlier than expected.
## Begin stochastic gradient ascent.
## iter ELBO delta_ELBO_mean delta_ELBO_med notes
## 100 -829.292 1.000 1.000
## 200 -817.856 0.507 1.000
## 300 -818.345 0.338 0.014
## 400 -816.956 0.254 0.014
## 500 -816.838 0.203 0.002 MEDIAN ELBO CONVERGED
## Drawing a sample of size 1000 from the approximate posterior...
## COMPLETED.
## Finished in 4.1 seconds.
##
## ************************************
## **** Model fitting is complete! ****
## ************************************Once the sampling is completed, all model-based regressors are
contained in the model_regressor list.
## store all subjects' stimulus value (SV) in ‘sv_all’
sv_all = output3$model_regressor$SV
dim(output3$model_regressor$SV) # number of rows=# of subjects (=10), number of columns=# of trials (=240)## [1] 10 240
## visualize SV (Subject #1)
plot(sv_all[1, ], type="l", xlab="Trial", ylab="Stimulus Value (subject #1)")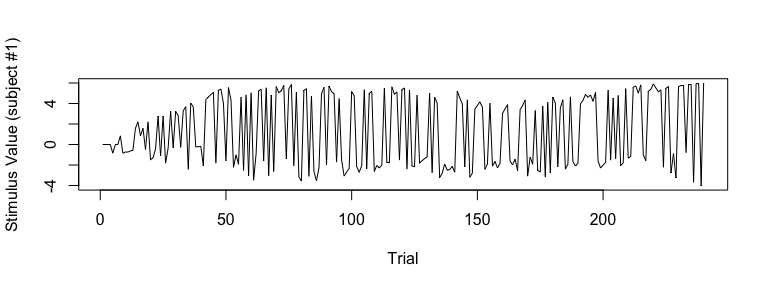
## visualize SV (Subject #5)
plot(sv_all[5, ], type="l", xlab="Trial", ylab="Stimulus Value (subject #5)")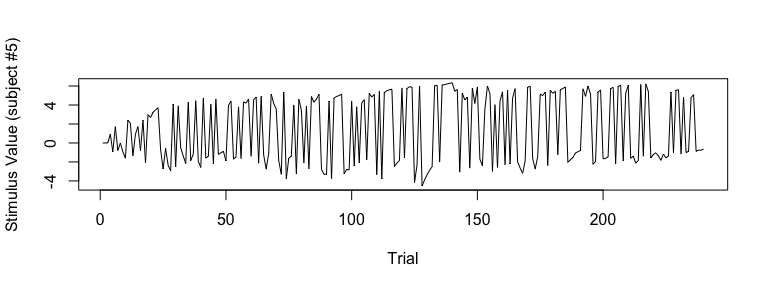
Similarly, users can extract and visualize other model-based
regressors. W(Go), W(NoGo),
Q(Go), Q(NoGo) are stored in
Wgo, Wnogo, Qgo, and
Qnogo, respectively.
6) Variational inference for approximate posterior sampling
Set vb = TRUE (default FALSE) to use Stan’s
variational algorithm —
cmdstanr::CmdStanModel$variational() under the hood — for
approximate posterior sampling. VB completes in seconds and is useful
for quick exploration, but final inferences should use MCMC.
To run gng_m3 with VB:
output3 = gng_m3(data="example", vb = TRUE)MCMC arguments (nchain, niter,
nthin, nwarmup) don’t apply when
vb = TRUE. The returned fit is a
CmdStanVB object instead of CmdStanMCMC. See
?cmdstanr::CmdStanModel$variational
for the underlying Stan options.
7) Posterior predictive checks
Simply put, posterior predictive checks refer to when a fitted model is used to generate simulated data and check if simulated data are similar to the actual data. Posterior predictive checks are useful in assessing if a model generates valid predictions.
From v0.5.0, users can run posterior predictive checks on all models
except drift-diffusion models in hBayesDM. Simulated data from posterior
predictive checks are contained in
hBayesDM_OUTPUT$par_vals$y_pred. In a future release, we
will include a function/command that can conveniently summarize and plot
posterior predictive checks. In the mean time, users can program their
own codes like the following:
## fit example data with the gng_m3 model and run posterior predictive checks
x = gng_m3(data="example", niter=2000, nwarmup=1000, nchain=4, ncore=4, inc_postpred = TRUE)
## dimension of x$par_vals$y_pred
dim(x$par_vals$y_pred) # y_pred --> 4000 (MCMC samples) x 10 (subjects) x 240 (trials)
[1] 4000 10 240
y_pred_mean = apply(x$par_vals$y_pred, c(2,3), mean) # average of 4000 MCMC samples
dim(y_pred_mean) # y_pred_mean --> 10 (subjects) x 240 (trials)
[1] 10 240
numSubjs = dim(x$all_ind_pars)[1] # number of subjects
subjList = unique(x$raw_data$subjID) # list of subject IDs
maxT = max(table(x$raw_data$subjID)) # maximum number of trials
true_y = array(NA, c(numSubjs, maxT)) # true data (`true_y`)
## true data for each subject
for (i in 1:numSubjs) {
tmpID = subjList[i]
tmpData = subset(x$raw_data, subjID == tmpID)
true_y[i, ] = tmpData$keyPressed # only for data with a 'choice' column
}
## Subject #1
plot(true_y[1, ], type="l", xlab="Trial", ylab="Choice (0 or 1)", yaxt="n")
lines(y_pred_mean[1,], col="red", lty=2)
axis(side=2, at = c(0,1) )
legend("bottomleft", legend=c("True", "PPC"), col=c("black", "red"), lty=1:2)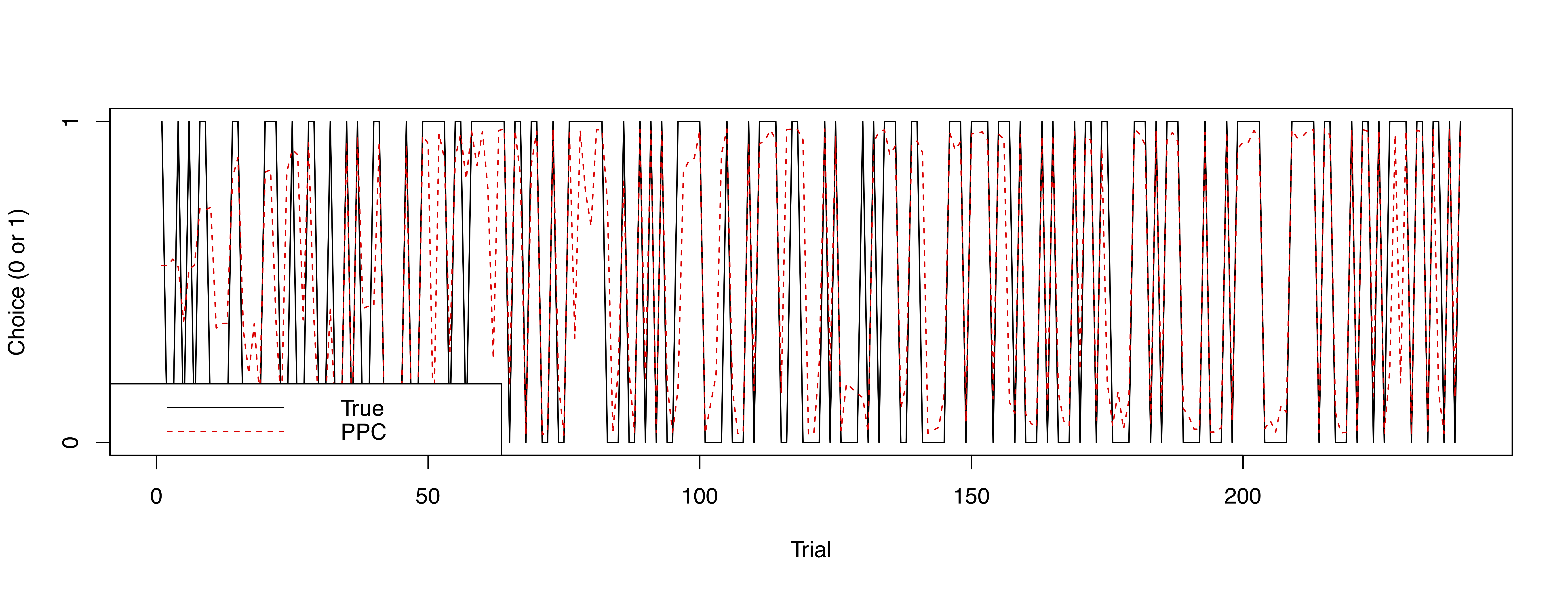
To-do list
We are planning to add more tasks/models. We plan to include the following tasks and/or models in the near future. If you have any requests for a specific task or a model, please let us know.
- More sequential sampling models (e.g., drift diffusion models with different drift rates for multiple conditions).
- Models for the passive avoidance learning task (Newman et al., 1985; Newman & Kosson, 1986).
- Models for the Stop Signal Task (SST)
- Allowing users to extract model-based regressors (O’Doherty et al., 2007) from more tasks.
Citation
If you used hBayesDM or some of its codes for your research, please cite this paper:
@article{hBayesDM,
title = {Revealing Neurocomputational Mechanisms of Reinforcement Learning and Decision-Making With the {hBayesDM} Package},
author = {Ahn, Woo-Young and Haines, Nathaniel and Zhang, Lei},
journal = {Computational Psychiatry},
year = {2017},
volume = {1},
pages = {24--57},
publisher = {MIT Press},
url = {doi:10.1162/CPSY_a_00002},
}Papers citing hBayesDM
Here is a selected list of papers that we know that used or cited hBayesDM (from Google Scholar). Let us know if you used hBayesDM for your papers!
Suggested reading
You can refer to other helpful review papers or books (Busemeyer & Diederich, 2010; Daw, 2011; Lee, 2011; Lee & Wagenmakers, 2014; Shiffrin et al., 2008) to know more about HBA or computational modeling in general.
Other Useful Links
- “Modelling behavioural data” by Quentin Huys, available in https://www.quentinhuys.com/teaching.html.
- Introductory tutorial on reinforcement learning by Jill O’Reilly and Hanneke den Ouden, available in http://hannekedenouden.ruhosting.nl/RLtutorial/Instructions.html.
- VBA Toolbox: A flexible modeling (MATLAB) toolbox using Variational Bayes (http://mbb-team.github.io/VBA-toolbox/).
- TAPAS: A collection of algorithms and software tools written in MATLAB. Developed by the Translational Neuromodeling Unit (TNU) at Zurich (http://www.translationalneuromodeling.org/tapas/).
- Bayesian analysis toolbox for delay discounting data, available in http://www.inferencelab.com/delay-discounting-analysis/.
- rtdists: Response time distributions in R, available in https://github.com/rtdists/rtdists/.
- RWiener: Wiener process distribution functions, available in https://cran.r-project.org/web/packages/RWiener/index.html.
Acknowledgement
This work was supported in part by the National Institute on Drug Abuse (NIDA) under award number R01DA021421 (PI: Jasmin Vassileva).